We have developed an efficient computational method that enables grand-canonical (GC) ab initio molecular dynamics (AIMD) simulations of the electrochemical system that tracks the dynamics of explicit solvent molecules in the presence of constant electrode potential. In particular, we have developed an SOLHYBRID model,1 an improvement of the implicit solvent model VASPSol,2, 3 that explicitly simulate the solvent, ions, adsorbates near the electrode while treating the solvent elsewhere implicitly. More importantly, to make application of the self-consistent grand canonical DFT (GC-DFT)4, 5 feasible for systems with few hundred atoms, we have recently developed the TPOT (Target POTential) routine1 that was implemented locally to the popular Vienna Ab initio Simulation Package (VASP).6, 7 It relies on an interactive optimization of the number of electrons for obtaining a predetermined target potential.1 Figure below shows an example of constant electrode potential AIMD simulation of the CO2 adsorption on Au(110) with the presence of K+ cation. It shows that the number of electrons in the simulation cell varies to keep the potential of the electrode constant. The results of the simulations show that CO2 forms a stable adsorption configuration with the presence of K+ at an electrode potential of -1V vs RHE. It is worth mentioning that CO2 is found to not adsorb on the Au(110) surface even with the presence of K+ in traditional DFT simulations.8
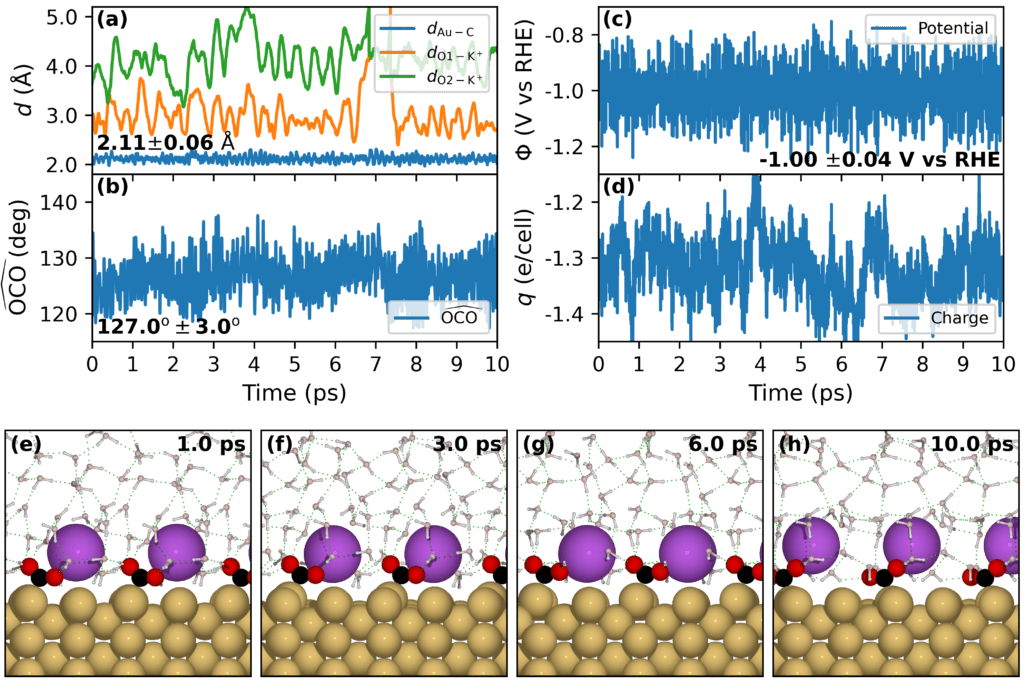
Details of this development can be found in the preprint.1 The TPOT package is available for free of charge at our GitHub.
Reference:
1. D. Le, “An Explicit-Implicit Hybrid Solvent Model for Grand Canonical Simulations of the Electrochemical Environment,” ChemRvix 10.26434/chemrxiv-2023-z2n4n 10.26434/chemrxiv-2023-z2n4n (2023). http://doi.org/10.26434/chemrxiv-2023-z2n4n
2. K. Mathew, V.S.C. Kolluru, S. Mula, S.N. Steinmann, and R.G. Hennig, “Implicit self-consistent electrolyte model in plane-wave density-functional theory,” The Journal of Chemical Physics 151, 234101 (2019). http://doi.org/10.1063/1.5132354
3. K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T.A. Arias, and R.G. Hennig, “Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways,” The Journal of Chemical Physics 140, 084106 (2014). http://doi.org/10.1063/1.4865107
4. R. Sundararaman, W.A. Goddard, and T.A. Arias, “Grand canonical electronic density-functional theory: Algorithms and applications to electrochemistry,” The Journal of Chemical Physics 146, 114104 (2017). http://doi.org/10.1063/1.4978411
5. R. Sundararaman and T.A. Arias, “Joint and grand-canonical density-functional theory” in “Atomic‐Scale Modelling of Electrochemical Systems”. (eds. M.M. Melander, T.T. LaurilaandK. Laasonen) 139-172 (John Wiley & Sons Ltd, 2021). http://doi.org/10.1002/9781119605652.ch4
6. J. Hafner, “Ab-initio simulations of materials using VASP: Density-functional theory and beyond,” Journal of Computational Chemistry 29, 2044-78 (2008). http://doi.org/10.1002/jcc.21057
7. G. Kresse and J. Furthmüller, “Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set,” Physical Review B 54, 11169-11186 (1996). http://doi.org/10.1103/PhysRevB.54.11169 8. X. Qin, T. Vegge, and H.A. Hansen, “CO2 activation at Au(110)–water interfaces: An ab initio molecular dynamics study,” The Journal of Chemical Physics 155, 134703 (2021). http://doi.org/10.1063/5.0066196