The activities of the Theoretical Attosecond Science group at UCF encompass a wide range of research topics associated to the theory, methodology, and phenomenology of the structure and dynamics of the electronic continuum in atoms and molecules, and of its interplay with coherent ultrashort light pulses. Our main interest is in the phenomenology induced by ultrashort pulses in an atomic or molecular target (which we refer to as ex situ spectroscopies, borrowing a term first introduced by Paul Corkum), with a special focus on the effect of many-body correlation and the role played by metastable states in ultrafast processes. Our main areas of research are
- Ab initio Theory of the Atomic Electronic Continuum
- Atomic Attosecond PhotoElectron Spectroscopy (APES)
- Atomic Attosecond Optical Spectroscopies (ATAS / 4WM)
- Double Photoionization
- Ab Initio Theory of the Molecular Electronic Continuum
- Vibrationally-Resolved Core Molecular Photoionization
- Molecular Attosecond Photoelectron and Optical Spectroscopies
Below, we offer a colloquial description and motivation for each of these areas, mainly directed to undergraduate and new graduate students, and list the PI’s pertinent publications.
Ab initio Theory of the Atomic Electronic Continuum
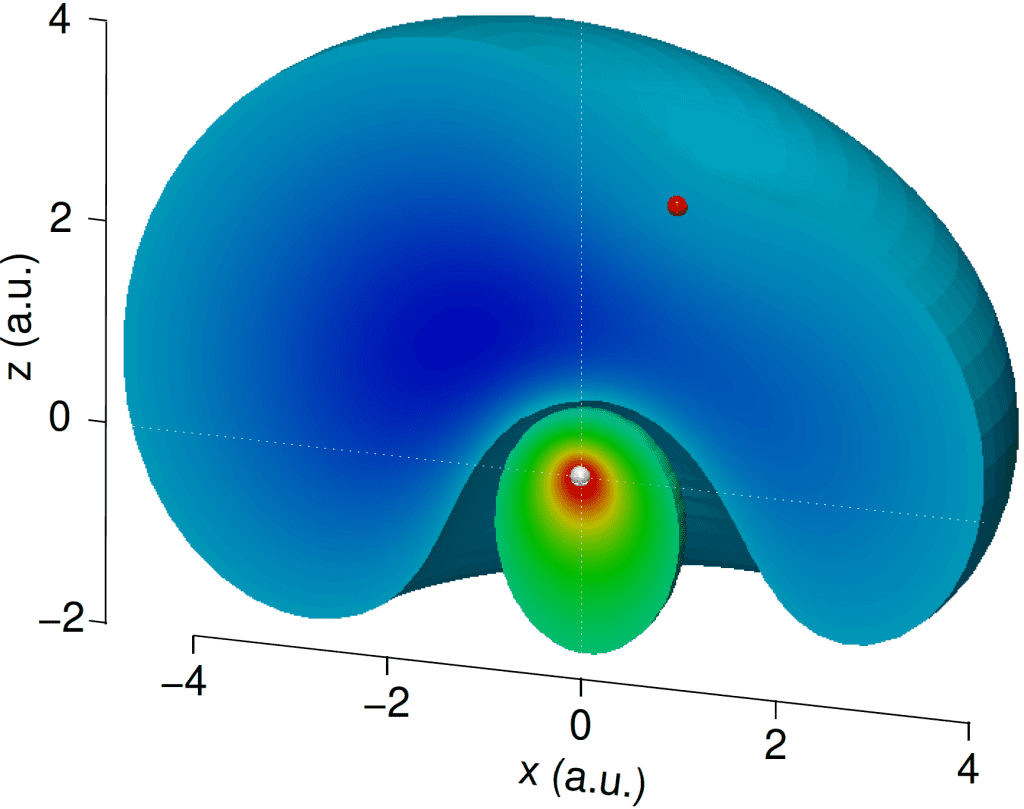
Atoms are ideal systems to study electronic correlation without the considerable geometric and dynamical complications brought about by the presence of multiple nuclei. Due to atoms’ rotational invariance, all the angular components of the many-body wave function can be treated analytically, which accelerates numerical calculations and allow us to achieve more precision than it is possible in molecules. Rare gases (He, Ne, Ar, Kr, Xe) are a darling of experimentalists because they are harmless, easy to handle, and they do not gunk up or corrode the equipment, so many experiments in attosecond science have rare gases as a target.
Our group has developed several programs to describe atomic ionization:
- HELIUM: a code that can treat a system of two-active electrons on top of a polarizable core. This was the code used to generate the picture on the left.
- TAEC: or Three-Active-Electrons Code, which can treat three electrons on top of a polarizable code, and has been used for accurate calculations of the resonant states of the boron atom
- NEWSTOCK: a general non-relativistic code for atomic ionization, which has been used to reproduce a number of photoelectrons and optical atomic observables.
All these programs are based on a method known as close-coupling, in which the configuration space for the system is expressed as a linear combination of products of ionic states and free-electron functions. Once this configuration space is complemented with a sufficiently rich set of localized configurations, to better reproduce electronic correlation in the neutral at short range, the close-coupling method is able to deliver accurate bound, resonant, and scattering states for the atom. These calculations require ad hoc codes to evaluate the matrix elements between close-coupling states, to solve the multi-channel Lippmann-Schwinger equation needed to determine the scattering states, and to isolate the states corresponding to autoionizing resonances through the imposition of appropriate boundary conditions. The group actively develops numerical methods to solve these problems in an effective manner.
Publications
- LA et al., Attosecond photoelectron spectroscopy of helium DES, PRR (2023).
- N Douguet et al., Application of the CK variational method to attosecond spectroscopy, PRA (2018).
- C Marante et al., Photoionization using the xchem approach: Total and partial cross sections of Ne […], PRA (2017).
- LA et al., Autoionizing states of atomic boron, PRA (2016).
- LA et al., PI of He by as pulses: Extraction of spectra from correlated wave functions, PRA (2013).
- T Carette et al., MCHF CC ansatz: Application to the Ar PICS and delays, PRA (2013).
- E Lindroth et al., Atomic resonance states and their role in charge-changing processes, AQC (2012).
- LA et al., Nondipole effects in helium photoionization, JPB (2010).
- LA et al., On the B-splines effective completeness, CPC (2009).
- LA, Rydberg and autoionizing triplet states in Helium up to the N= 5 threshold, ADNDT (2008).
- LA et al., Helium 23S photoionization up to the N= 5 threshold, JPB (2008).
- LA et al., 3S, 3Po,e, 3De,o resonance series in helium, JPB (2007).
- LA et al., He photoionization: βN and σN below N = 5 and 6 thresholds, TCA (2007).
- LA et al., K-matrix method with B-splines: σnℓ, βn and resonances in He PI below N= 4 threshold, JPB (2006).
Atomic Attosecond PhotoElectron Spectroscopy (APES)
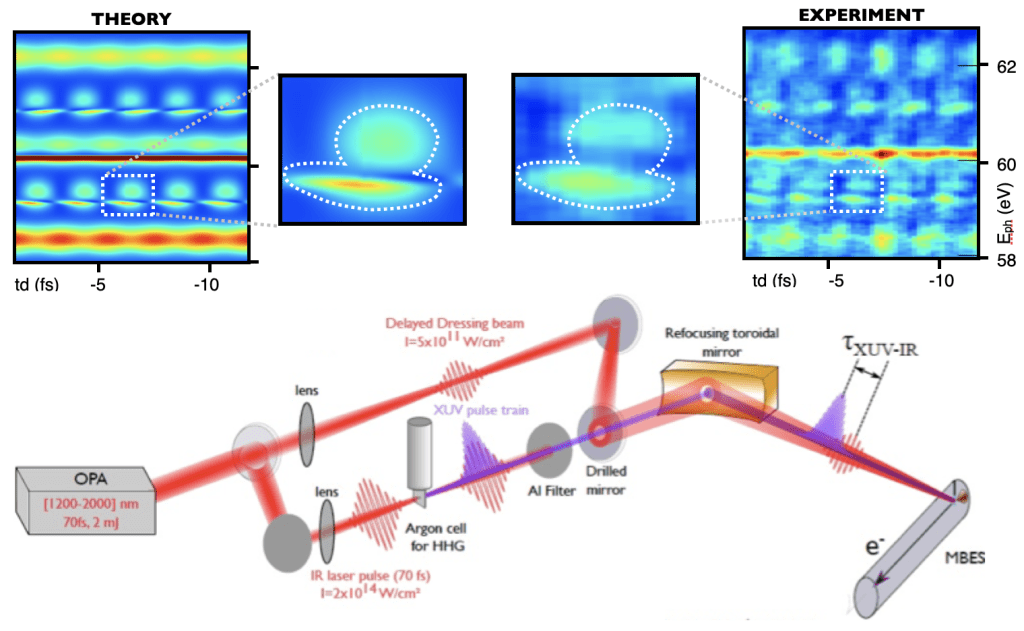
Attosecond pulses typically trigger the emission of a photoelectron. The probability with which a photoelectron is emitted in a certain range of energies and angles (photoelectron distribution) carries information on the state of the target when the ionizing pulse invested it. It is no surprise, therefore, that the detection of photoelectrons is one of the main tools used by physicists in attosecond experiments. In contrast to traditional linear photoelectron spectroscopy, attosecond spectroscopies rely on the exchange of multiple photons from pulses with variable temporal delays to activate alternative quantum paths that can interfere. For example, if an atom absorbs a 60eV photon and 1.5eV photon, the result is the same as if it absorbs a 63eV photon and emits a 1.5eV photon. In attosecond spectroscopies, all these photons are present at the same time, with stable relative phases. As a result, both quantum paths are active and they can interfere destructively or constructively. From this interference, it is possible to determine the relative phase between the quantum paths and reconstruct the delay with which an electron is emitted to the continuum. This is the principle at the basis of RABBITT (Reconstruction of Attosecond Beating By Interference of Two-photon Transitions), one of the most widely used techniques in attosecond science.
Publications
- LA et al., Attosecond photoelectron spectroscopy of helium doubly excited states, PRR (2023).
- S Mehmood et al., Ionic coherence in resonant above-threshold attosecond ionization spectroscopy, PRA (2023).
- D Busto et al., Probing electronic decoherence with high-resolution attosecond photoelectron interferometry, EPJD (2022).
- S Mehmood et al., Coherence control in helium-ion ensembles, PRR (2021).
- J Fuchs et al., Towards the complete phase profiling of attosecond wave packets, PRR (2021).
- M Turconi et al., Spin–orbit-resolved spectral phase measurements around a Fano resonance, JPB (2020).
- J Fuchs et al., Time delays from one-photon transitions in the continuum, Optica (2020).
- S Donsa et al., Circular holographic ionization-phase meter, PRL (2019).
- L Barreau et al., Disentangling spectral phases of interfering AI states from as interferometric measurements, PRL (2019).
- N Douguet et al., Application of the complex Kohn variational method to attosecond spectroscopy, PRA (2018).
- C Cirelli et al., Anisotropic photoemission time delays close to a Fano resonance, Nature Commun (2018).
- D Busto et al., Time–frequency representation of autoionization dynamics in helium, JPB (2018).
- LA et al., Control of photoemission delay in resonant two-photon transitions, PRA(2017).
- S Heuser et al., Angular dependence of photoemission time delay in helium, PRA (2016).
- V Gruson et al., Attosecond dynamics through a Fano resonance: Monitoring the birth of a photoelectron, Science (2016).
- Á Jiménez-Galán et al., Two-photon finite-pulse model for resonant transitions in attosecond experiments, PRA (2016).
- M Kotur et al., Spectral phase measurement of a Fano resonance using tunable attosecond pulses, Nature Commun (2016).
- Á Jiménez-Galán et al., Modulation of attosecond beating in resonant two-photon ionization, PRL (2014).
- Á Jiménez Galán et al., The soft-photon approx. in IR-assisted atomic ionization by XUV APTs, NJP (2013).
- LA et al., Photoionization of helium by as pulses: Extraction of spectra from correlated wave functions, PRA (2013).
- LA et al., Ionization branching ratio control with a resonance attosecond clock, PRL (2010).
Atomic Attosecond Optical Spectroscopies
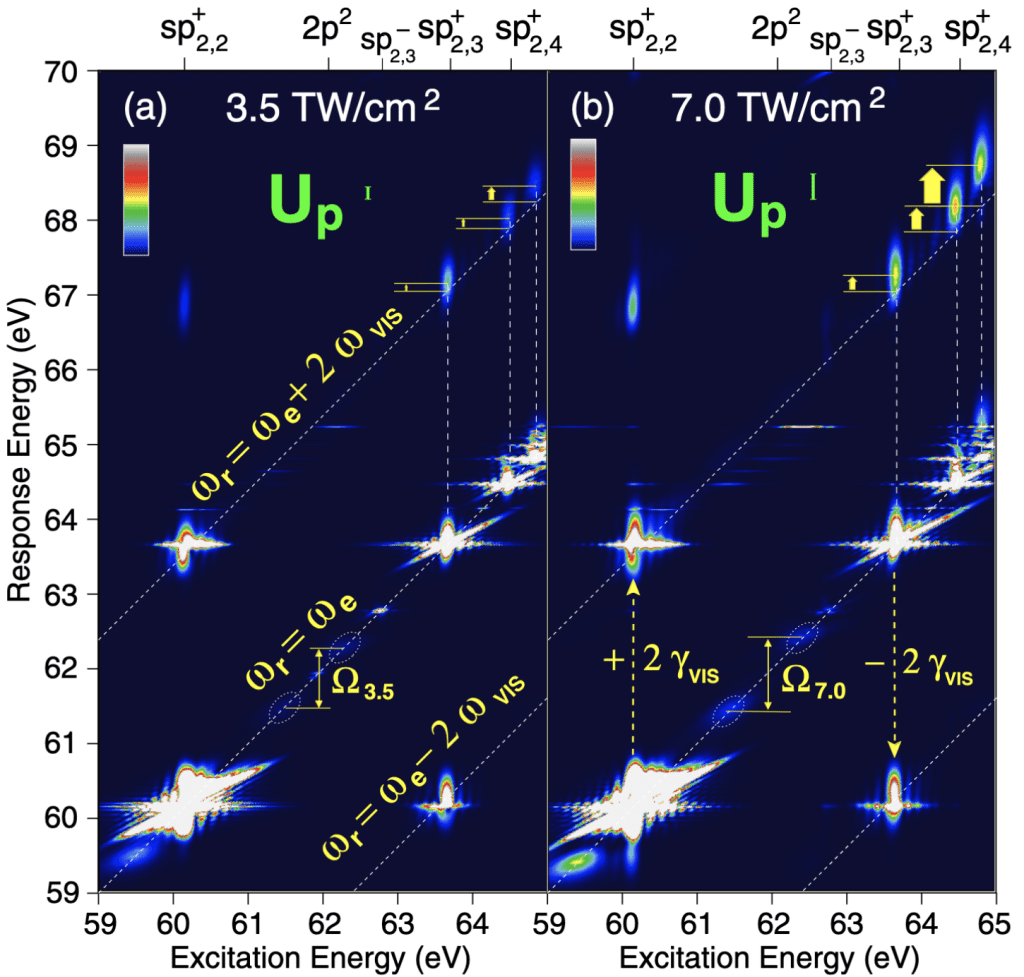
The interaction between an attosecond pulse and a target atom or molecule does not alter only the target, it also alters the light transmitted through the sample. Indeed, all the particles invested by the light develop oscillating electric dipole moments that radiate coherently at their oscillation frequency. This secondary emission can interfere with the incident radiation. In the case of linear spectroscopies, this interference is destructive, leading to light absorption. In non-linear spectroscopies such as Attosecond Transient Absorption Spectroscopy (ATAS), on the other hand, the interference can be constructive. In Four Wave Mixing Spectroscopy (FWMS), when the pump and probe pulses are not collinear, induced dipoles generally emit light in different directions, so the incident and secondary light do not interfere at all (background-free signal).
In a pump-probe setup, the change in the spectrum of an attosecond light pulse transmitted through a sample as a function of the relative delays between the pulses in the excitation sequence contains valuable information on the ultrafast dynamics in the target. As it turns out, most of the optical response of a system originates from the immediate neighborhood of the target atom or molecule. Furthermore, to a good approximation, the generated radiation propagates freely to the detector. Optical spectroscopies, therefore, give more direct access to the target dynamics than photoelectron spectroscopies do, and they are also considerably easier to simulate theoretically, since they do not require to keep track of the fate of the emitted photoelectrons. As soon as a photoelectron leaves the molecular region and it can be safely assumed to never come back, it can be excised from the simulation.
Another distinctive feature of optical methods is that the target dipole is dominated by the beating of the excited component of the wave function with the ground-state component, which is normally preponderant. As a result, optical methods single out only the states of the excited wave function whose transition to the ground state is allowed by dipole selection rules. While this circumstance may appear a limit, it is in fact a feature: simpler spectra mean cleaner signals that can be more easily interpreted.
Publications
- N G Puskar et al., Measuring autoionization decay lifetimes of optically forbidden inner valence excited states in neon atoms with attosecond noncollinear four-wave-mixing spectroscopy, PRA (2023).
- V Leshchenko et al., Kramers–Kronig relation in attosecond transient absorption spectroscopy, Optica (2023).
- S Yanez-Pagans et al., Multipolariton control in attosecond transient absorption of autoionizing states, PRA (2022).
- N Harkema et al., Autoionizing polaritons in attosecond atomic ionization, PRL (2021).
- A Chew et al., Attosecond transient absorption spectrum of argon at the L2,3 edge, PRA (2018).
- CLM Petersson et al., Attosecond transient absorption spectroscopy of helium above the ionization threshold, PRA (2017).
- LA et al., Dressing effects in the attosecond transient absorption spectra of doubly excited states in helium, PRA (2015).
- C Ott et al., Reconstruction and control of a time-dependent two-electron wave packet, Nature (2014).
Double Photoionization
In double photoionization processes, one or more photons eject two electrons from a target, instead of just one
One-photon double ionization strongly depends on electron correlation, which is a quantity of primary importance in determining the properties of atoms and molecules. In a pump-probe setting, the emission of a second electron by means of the absorption of multiple photons allows us to directly image, as a function of time, the reaction of the residual electrons in the target after the first photoelectron has been emitted. In single-ionization spectroscopy, this reaction can only indirectly be reconstructed. If two photons are absorbed in sequence, and each absorption is associated with the emission of a photoelectron, we say the process occurs in the sequential regime, i.e., two single ionizations taking place one after the other: simple enough. If the two photons are absorbed very close to each other, however, or if the photon energy is sufficient to eject an electron from the neutral target but not from the resulting charged ion, a more complex non-sequential regime sets in.
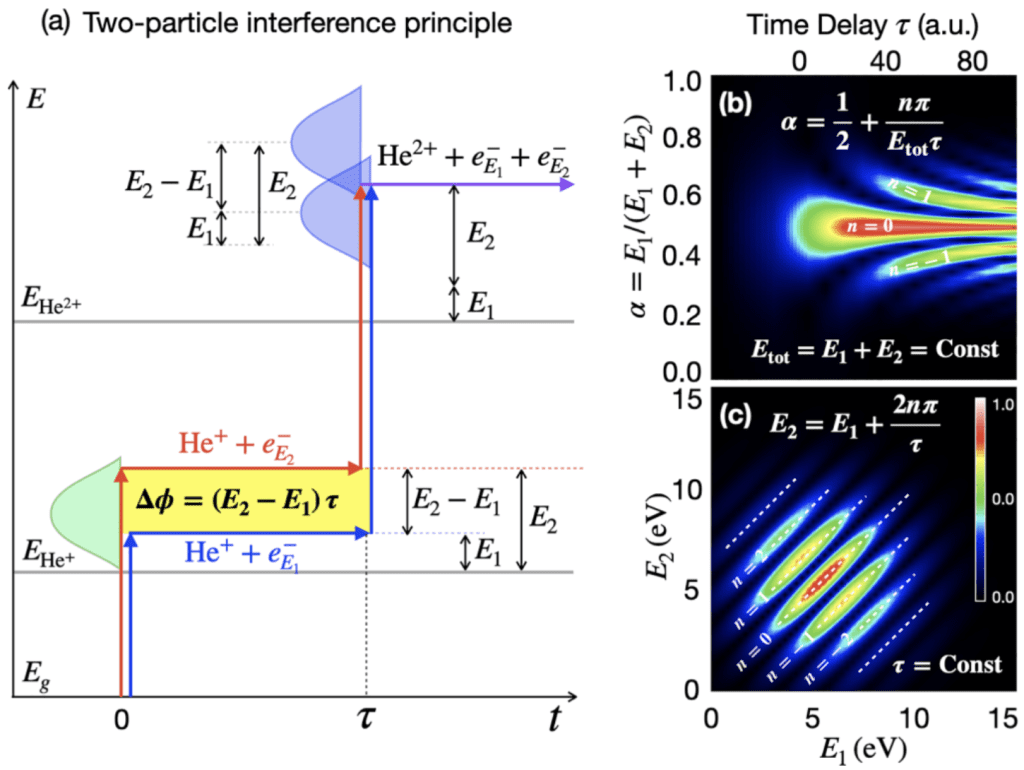
The accurate ab initio theoretical description of a general double ionization process is a devilishly hard problem, due to its dimensionality (as a rule of thumb, think a thousand times more computationally expensive than simulating single-ionization processes). Yet, the prize is valuable enough to be worth the effort. Furthermore, if one does not try to reproduce the direction photoelectrons are emitted, there are promising approximated alternatives to the direct solution of the time-dependent Schrödinger equation that can give reasonable results at fraction of the computational cost.
Publications
- S Chattopadhyay et al., TPDI with finite pulses: Application of the virtual sequential model to He, PRA (2023).
- LA et al., Two-Particle Coulomb Green Function Method with Projected Potential: Application to He DI, JPCA (2009).
- LA et al., Photo–double-ionization of the n s shell of rare gases, PRA (2009).
- LA et al., A general algorithm for fitting efficiently triple differential cross sections of atomic DI, JPB (2008).
Ab Initio Theory of the Molecular Electronic Continuum
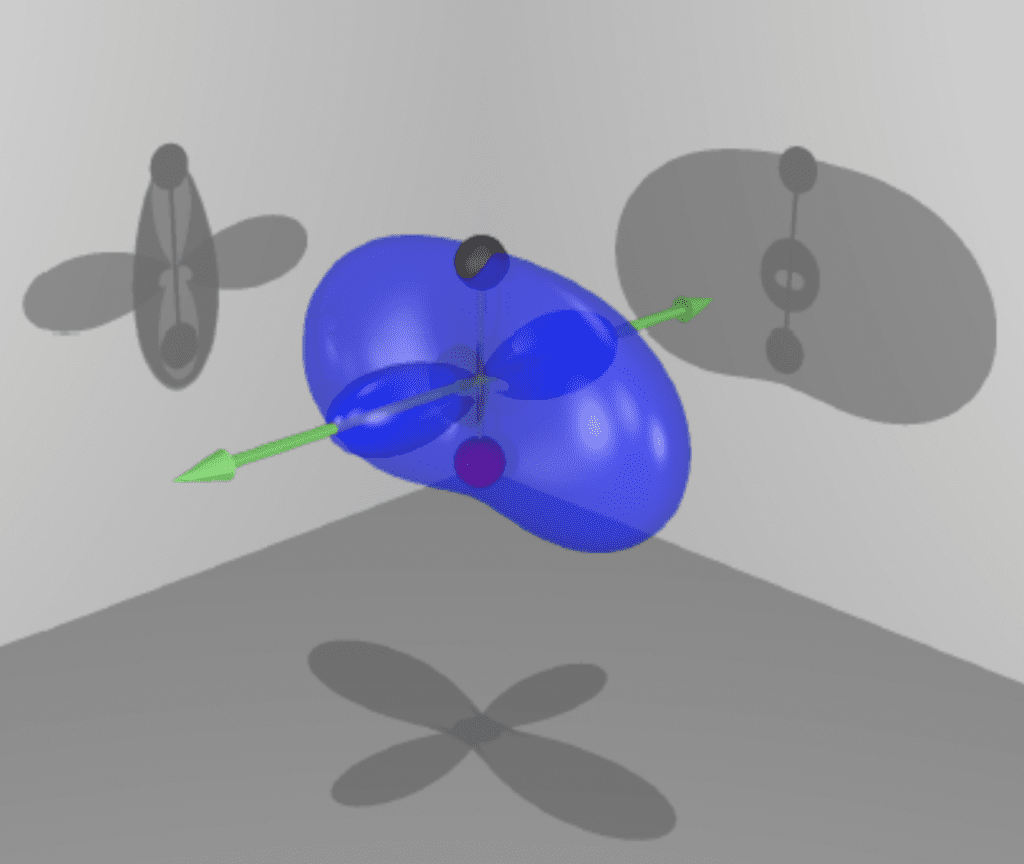
Molecules differ from atoms in that they have several nuclei instead of just one. This circumstance is accompanied by several theoretical challenges. Once one gives up a single center where the positive charge is located, the symmetry of the system is drastically reduced, the separation between angular and radial degrees of freedom is lost, and so is the great simplification that spherical symmetry affords us in atoms. A reliable structure code for the electronic continuum within the fixed-nuclei approximation is a necessary prerequisite to any molecular calculation.
Nuclei, of course, are not static. While they normally move slowly compared to electrons, electrons occasionally move as slowly as the nuclei. When that happens, the nuclear and the electronic degrees of freedom become entangled. These so-called non-adiabatic couplings occur when the electronic potential energy surfaces in Born-Oppenheimer approximation are close to each other. In attosecond science, which concerns itself with the electronic continuum, these complications are compounded by the challenges inherent to the description of molecular scattering wave functions.
One of the main activities in our group is the development of ASTRA (AttoSecond TRAnsitions), a new molecular ionization code based on the efficient formalism of transition density matrices. This research thrust started in 2019, thanks to the support of the Department of Energy, and it benefits from an ongoing and strong collaboration with Barry I Schneider, at the National Institute of Standard and Technology in Gaithersburg, Maryland, and with Jeppe Olsen, at the Chemistry Department of Århus University, Denmark.
We also collaborate with Fernando Martín’s group at the Autonomous University of Madrid to develop XCHEM, a molecular ionization code whose development started in 2012, and with Nicolas Douguet (soon at UCF!), to an extension of the Complex Kohn – MESA code to two-photon processes.
Publications
- J M Randazzo et al., ASTRA a Transition-Density-Matrix Approach to Molecular Ionization, PRR (2023).
- V J Borràset al., MFPADs of CO in the vicinity of Feshbach resonances: An XCHEM approach, JCTC (2021).
- H Gharibnejad et al., A multi-center quadrature scheme for the molecular continuum, CPC (2021).
- N Douguet et al., Application of the complex Kohn variational method to attosecond spectroscopy, PRA (2018).
- M Klinker et al., Partial cross sections and interfering resonances in PI of N2, PRA (2018).
- M Klinker et al., Electron Correlation in the Ionization Continuum of Molecules: PI of N2 in […], JPCL (2018).
- M Waitz et al., Imaging the square of the correlated two-electron wave function of H2, Nature Commun (2017).
- C Marante et al., Hybrid-basis close-coupling interface to QC packages for the treatment of ionization problems, JCTC (2017).
- C Marante et al., Hybrid Gaussian–B-spline basis for the electronic continuum: PI of atomic hydrogen, PRA (2014).
Vibrationally-Resolved Core Molecular Photoionization
The ionization of molecules from core orbitals has several unique features that distinguish it from valence ionization. Core orbitals are typically highly localized on individual atoms. As a result, their excitation is highly sensitive to the chemical environment local to that atom. Since the photoelectron emerges from a well-defined center, diffraction from nearby atoms appears in the photoelectron angular distribution of pre-oriented molecules, allowing one to reconstruct their geometry. Core ionization is generally followed by Auger decay: one electron from the valence or inner valence fills the vacancy in the core orbital and the excess energy thus liberated is absorbed by another electron which leaves the molecule. Auger decay is an extremely interesting process because it informs us on the correlation between the pair of electrons that participate in it. The scattering of the photoelectron from nearby atoms manifests itself also in the integral cross section as EXAFS like oscillations, which also incorporate information on the molecular structure (see figure on the side).
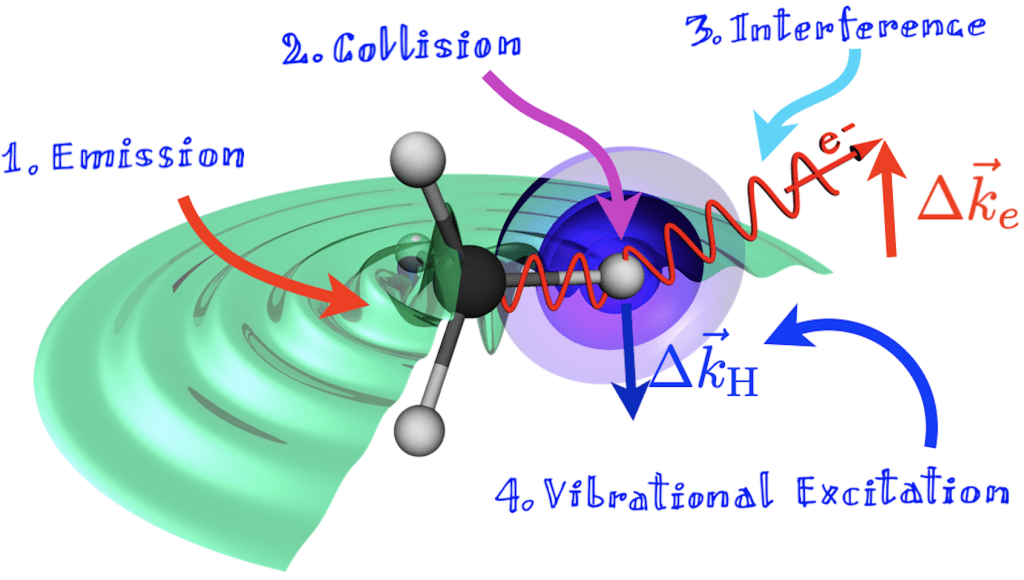
In molecules that contain tightly bound identical atoms from the second period, such as acetylene and molecular nitrogen, both of which feature strong triple bonds, core orbitals marginally overlap giving rise to a splitting comparable to or larger than the Auger lifetime of a core hole. As a result, core ionization can give rise to a vacancy that is delocalized on two atoms. Since photoelectron emission occurs synchronously from two nearby sites, the photoelectron angular and energy distribution feature the characteristic interference fringes of the famous Young’s double slit experiment. In molecules, this phenomenon is known as Cohen-Fano interference.
All these oscillations are normally superimposed on a cross section that decays rapidly with energy, so their determination has a fair degree of uncertainty, since it presupposes that one knows what the ionization cross section would be in absence of interference (which one doesn’t, really!). This problem can be circumvented by noticing that both photoemission and intramolecular photoelectron scattering require that the emitting or scattering center recoil to preserve momentum: the nuclei receive a kick. As a result, the molecule gets vibrationally excited. Since both emission and scattering are susceptible to interference, so is the vibrational excitation. If the energy resolution of the light source is sufficiently high, and if the separation between consecutive vibrational states of the core-hole molecule is larger than the natural width Auger decay imparts to the state, then it is possible to separately measure the vibrationally resolved peaks in the photoelectron spectrum. While the amplitude of all these peaks decays rapidly with photoelectron energy, their ratio does not. Instead, it encodes in beautiful oscillatory patterns all the interference phenomena mentioned above.
Publications
- B Ghomashi et al., Attosecond intramolecular scattering and vibronic delays, PRL (2021).
- D Ayuso et al., Vibrationally Resolved B 1s Photoionization Cross Section of BF3, JPCA (2015).
- M Patanen et al., Vibrationally resolved C 1s photoionization cross section of CF4, JPB (2014).
- K Ueda et al., Intramolecular photoelectron diffraction in the gas phase, JCP (2013).
- E Kukk et al., Effects of molecular potential and geometry on atomic core-level photoemission […], PRA (2013).
- LA et al., Double-slit experiment with a polyatomic molecule: vibrationally resolved C 1s PES of acetylene, NJP (2012).
- E Plésiat et al., Intramolecular electron diffraction in vibrationally resolved K-shell photoionization of methane, PRA (2012).
Molecular Attosecond Photoelectron and Optical Spectroscopy
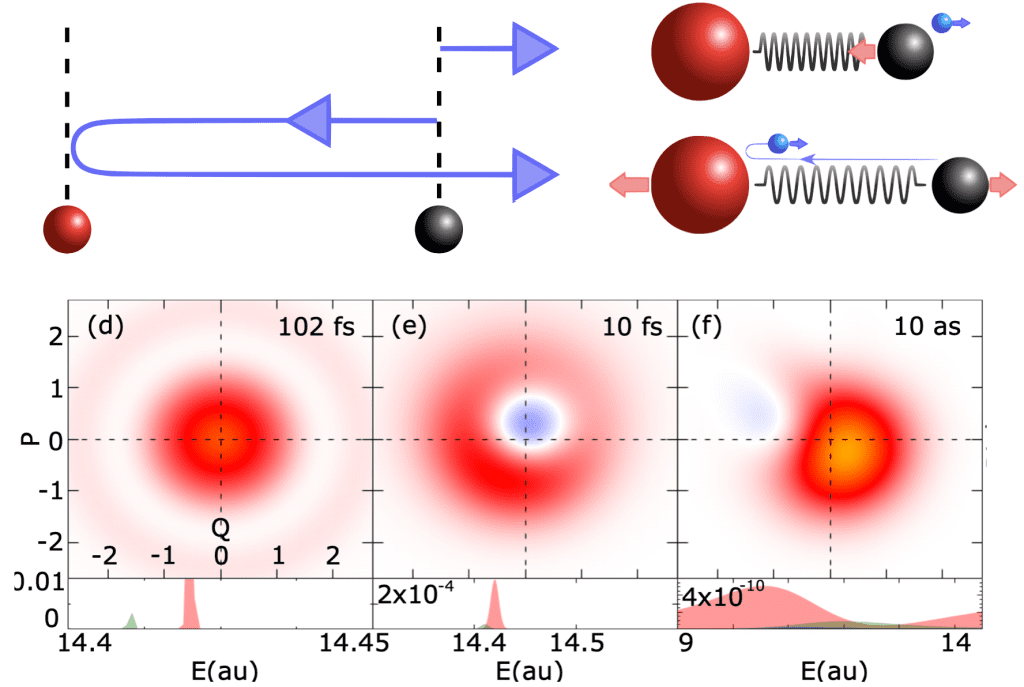
As attosecond science matures, its focus is shifting from proof-of-principle experiments in elementary systems to the study of ultrafast transformations in molecules of chemical and even biological interest. Elucidating the role of electronic correlation and entanglement in these processes requires many-body tools such as molecular close-coupling methods or multi-reference time-dependent self-consistent-field methods. As mentioned above, our group works at the development of close-coupling methods, such as ASTRA and XCHEM, which offer advantages in the asymptotic representation of the photoelectron wave packets.
A particularly interesting phenomenon is charge migration: an attosecond pulse, or a short intense infrared pulse ionizes a target molecule giving rise to a non-stationary localized vacancy, which wanders across the molecule. The interest in this phenomenon is due to the possibility of using it to transmit charge at high speed, as well as to localize, by means of a second pulse, the charge on a specific moiety, thus altering the molecular reactivity altogether.
Over time, the picture of charge migration provided by calculations in fixed-nuclei approximation becomes less and less reliable. Since electronic and nuclear motions are entangled, the separate motion of either becomes blurred within a handful of femtoseconds, a process known as decoherence. While the molecule as a whole may remain in a well-defined quantum-mechanical state, it is difficult to monitor and control multiple degrees of freedom at the same time. For practical purposes, therefore, the original quantum coherence is effectively lost. In fact, if the ultrafast process starts with an ionization event, the emerging ion and the photoelectron are entangled at the outset. Unless the quantum states of the ion and of the photoelectron are detected in coincidence, therefore, the molecular ion is only partly coherent from the get-go.
Things become even more interesting when one considers non-adiabatic effects, which affect the motion of nuclear wave packets in proximity of tightly avoided crossings, conical intersection seams, and exceptional points. Until recently, tackling theoretically these processes in the electronic continuum from first principles would not only be a hard proposition, but also one destined to be frustrated, since these processes could not be observed experimentally with the required temporal resolution anyway. With the advent and maturation of attosecond technology, however, the proposition remains hard, but at least it becomes worth the effort.
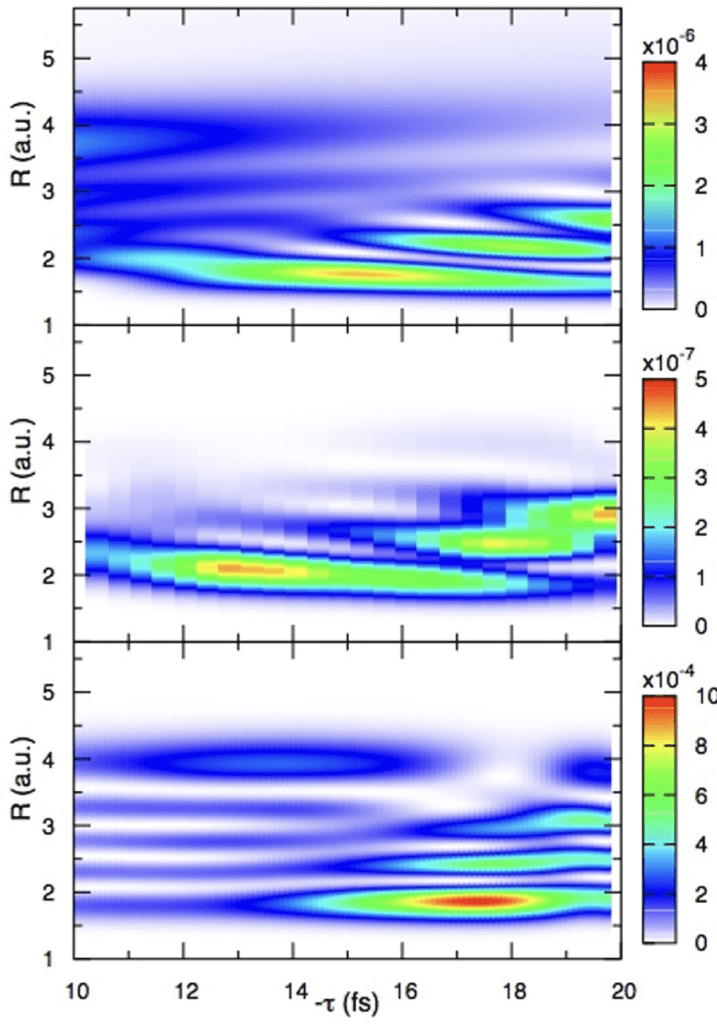
Publications
- Vicent J Borràs, Jesús González-Vázquez, Luca Argenti, Fernando Martín, Attosecond photoionization delays in the vicinity of molecular Feshbach resonances, Science Advances 9, eade3855 (2023).
- Bejan Ghomashi, Nicolas Douguet, Luca Argenti, Attosecond intramolecular scattering and vibronic delays, Phys. Rev. Lett. 127, 203201 (2021).
- N Saito et al., Attosecond electronic dynamics of core-excited states of in the soft x-ray region, PRR (2021).
- Y Cheng et al., Reconstruction of an excited-state molecular wave packet with ATAS, PRA (2016).