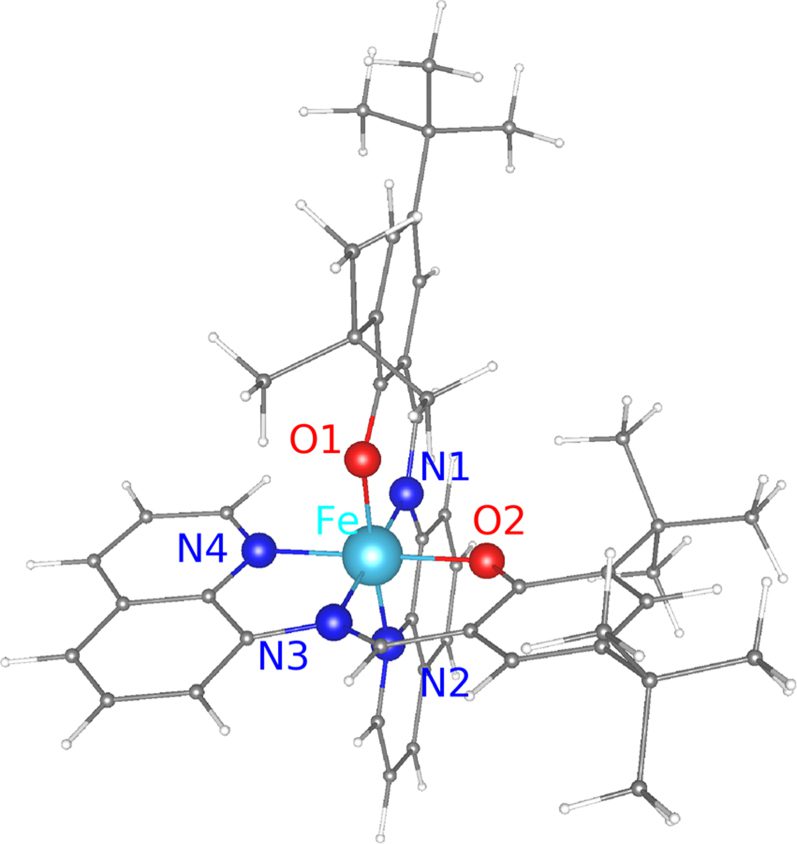
Au(111) is one of the substrates often used for supporting spin crossover (SCO) molecules, partly because of its inertness and partly because it is conducting. Using density functional theory based calculations of [Fe(tBu2qsal)2] SCO molecules adsorbed on the Au(111) surface, we show that while Au(111) may not be a suitable support for the molecule, it may be so for a monolayer (ML) of molecules. While, physisorption of [Fe(tBu2qsal)2] on Au(111) leads to electron transfer from the highest occupied molecular orbital to the substrate, electron transfer is minimal for a ML of [Fe(tBu2qsal)2] on Au(111), causing only negligible changes in the electronic structure and magnetic moment of the molecules. Furthermore, a small difference in energy between the ferromagnetic and antiferromagnetic configurations of the molecules in the ML indicates a weak magnetic coupling between the molecules. These results suggest Au(111) as a plausible support for a ML of [Fe(tBu2qsal)2], making such a molecular assembly suitable for electronic and spin transport applications. As for [Fe(tBu2qsal)2] SCO molecules themselves, we find hexagonal boron nitride (h-BN) to be a viable support for them, as there is hardly any charge transfer, while graphene displays stronger interaction with the molecule (than h-BN does) resulting in charge transfer from the molecule to graphene.
This work was published in Journal of Physics: Condensed Matter.
[D. Le, T. Jiang, M. Gakiya-Teruya, M. Shatruk, and T. S. Rahman, “On stabilizing spin crossover molecule [Fe(tBu2qsal)2] on suitable supports: insights from ab initio studies,” Journal of Physics: Condensed Matter 33, 385201 (2021). https://doi.org/10.1088/1361-648x/ac0beb]