Talat S. Rahman
UCF Trustee Chair Professor and Pegasus Professor
Department of Physics
University of Central Florida
- Multi-scale modeling of chemical reactions and related phenomena at surfaces
- Understanding processes that control growth and morphological evolution of thin films
- Theory and modeling of vibrational, optical and magnetic properties of nanomaterials
- Predictive modeling of functional two-dimensional transition metal dichalcogenides
- Surface coordination chemistry: novel functionality via substrate charge transfer and oxidation state
- Understanding the response of surfaces and nanostructures to ultrafast external fields
- Development of techniques beyond density functional theory for strongly correlated material
- Development of techniques suitable for non-equilibrium phenomena and non-adiabatic processes
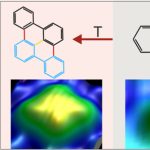
Increased Selectivity in Photolytic Activation of Nanoassemblies Compared to Thermal Activation in On-Surface Ullmann Coupling
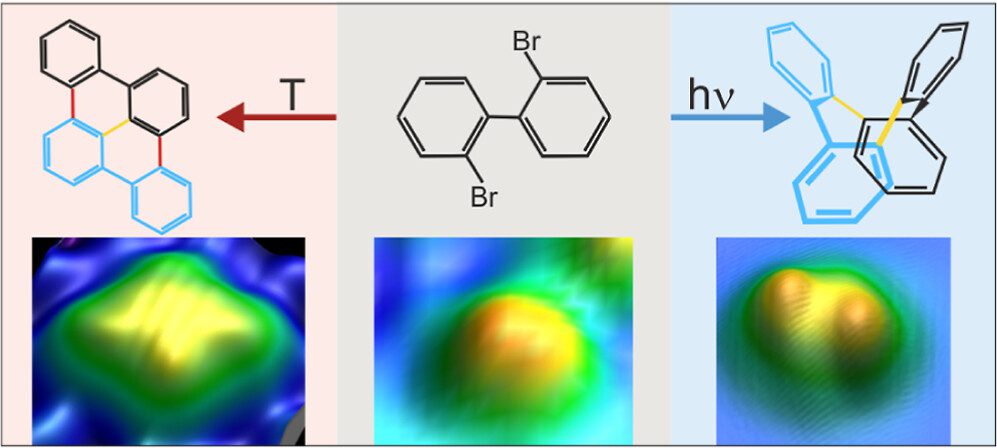
On-surface synthesis is a powerful method that has emerged recently to fabricate a large variety of atomically precise nanomaterials on surfaces based on polymerization. It is very successful for thermally activated reactions within the framework of heterogeneous catalysis. As a result, it often lacks selectivity. We propose to use selective activation of specific bonds as a crucial ingredient to synthesize desired molecules with high selectivity. In this approach, thermally nonaccessible products are expected to arise in photolytically activated on-surface reactions with high selectivity. We demonstrate for assembled 2,2′-dibromo biphenyl clusters on Cu(111) that the thermal and photolytic activations yield distinctly different products, combining submolecular resolution of individual product molecules in real-space imaging by scanning tunneling microscopy with chemical identification in X-ray photoelectron spectroscopy and supported by ab initio calculations. The photolytically activated Ullmann coupling of 2,2′-dibromo biphenyl is highly selective, with only one identified product. It starkly contrasts the thermal reaction, which yields various products because alternate pathways are activated at the reaction temperature. Our study extends on-surface synthesis to a directed formation of thermally inaccessible products by direct bond activation. It promises tailored reactions of nanomaterials within the framework of on-surface synthesis based on the photolytic activation of specific bonds.
This work was published in ACS Nano.
[C. Schunke, P. Schweer, E. Engelage, D. Austin, E. D. Switzer, T. S. Rahman, K. Morgenstern, “Increased Selectivity in Photolytic Activation of Nanoassemblies Compared to Thermal Activation in On-Surface Ullmann Coupling.” ACS Nano (2024). https://doi.org/10.1021/acsnano.3c11509]
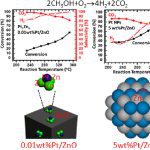
Breaking Continuously Packed Bimetallic Sites to Singly Dispersed on Nonmetallic Support for Efficient Hydrogen Production
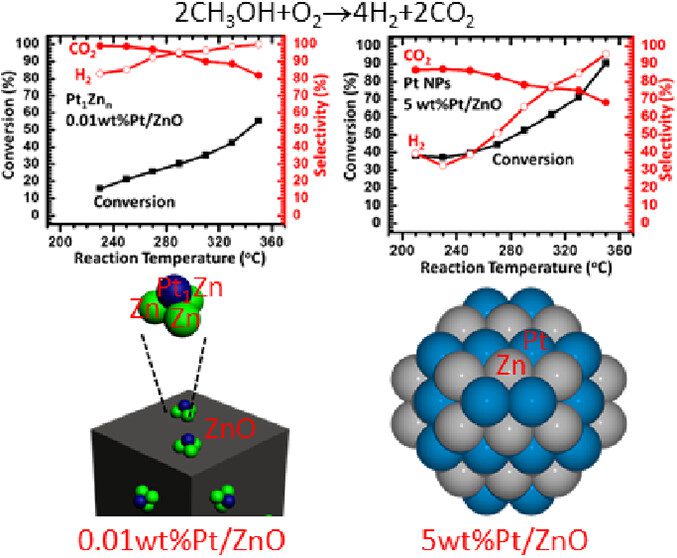
We have synthesized Pt1Zn3/ZnO, also termed 0.01 wt %Pt/ZnO-O2–H2, as a catalyst containing singly dispersed single-atom bimetallic sites, also called a catalyst of singly dispersed bimetallic sites or a catalyst of isolated single-atom bimetallic sites. Its catalytic activity in partial oxidation of methanol to hydrogen at 290 °C is found to be 2–3 orders of magnitude higher than that of Pt–Zn bimetallic nanoparticles supported on ZnO, 5.0 wt %Pt/ZnO-N2–H2. Selectivity for H2 on Pt1Zn3/ZnO reaches 96%–100% at 290–330 °C, arising from the uniform coordination environment of single-atom Pt1 in singly dispersed single-atom bimetallic sites, Pt1Zn3 on 0.01 wt %Pt/ZnO-O2–H2, which is sharply different from various coordination environments of Pt atoms in coexisting PtxZny (x ≥ 0, y ≥ 0) sites on Pt–Zn bimetallic nanoparticles. Computational simulations attribute the extraordinary catalytic performance of Pt1Zn3/ZnO to the stronger adsorption of methanol and the lower activation barriers in O–H dissociation of CH3OH, C–H dissociations of CH2O to CO, and coupling of intermediate CO with atomic oxygen to form CO2 on Pt1Zn3/ZnO as compared to those on Pt–Zn bimetallic nanoparticles. It demonstrates that anchoring uniform, isolated single-atom bimetallic sites, also called singly dispersed bimetallic sites on a nonmetallic support can create new catalysts for certain types of reactions with much higher activity and selectivity in contrast to bimetallic nanoparticle catalysts with coexisting, various metallic sites MxAy (x ≥ 0, y ≥ 0). As these single-atom bimetallic sites are cationic and anchored on a nonmetallic support, the catalyst of singly dispersed single-atom bimetallic sites is different from a single-atom alloy nanoparticle catalyst. The critical role of the 0.01 wt %Pt in the extraordinary catalytic performance calls on fundamental studies of the profound role of a trace amount of a metal in heterogeneous catalysis.
This work was published in ACS Applied Materials & Interfaces.
[T. Jiang, Y. Li, Y. Tang, S. Zhang, D. Le, T. S. Rahman, F. Tao, “Breaking Continuously Packed Bimetallic Sites to Singly Dispersed on Nonmetallic Support for Efficient Hydrogen Production.” ACS Appl. Mater. Interfaces, 16, 21757 (2024). https://doi.org/10.1021/acsami.3c18160]
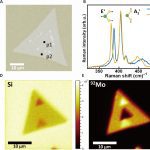
Anomalous isotope effect on the optical bandgap in a monolayer transition metal dichalcogenide semiconductor
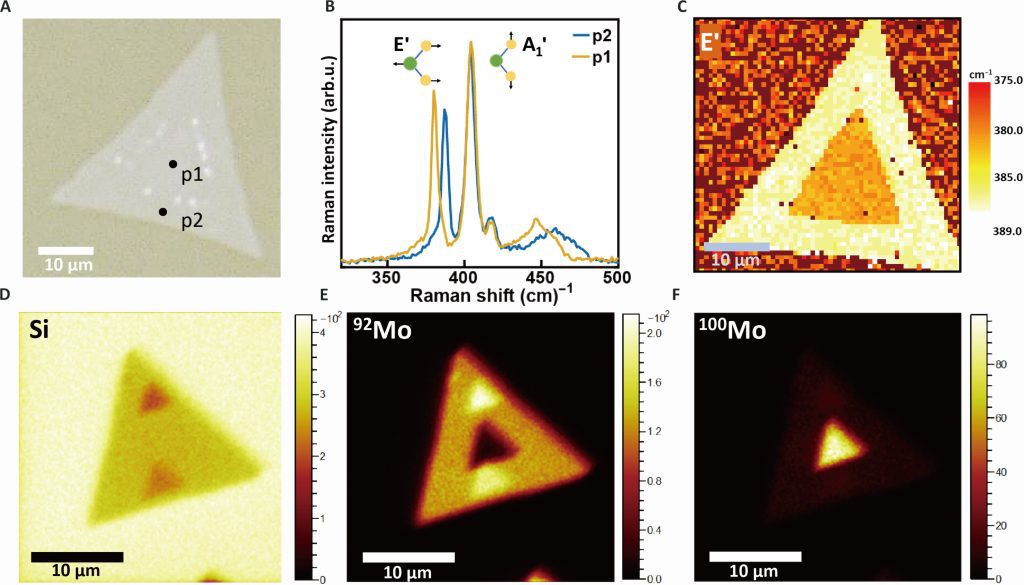
Isotope effects have received increasing attention in materials science and engineering because altering isotopes directly affects phonons, which can affect both thermal properties and optoelectronic properties of conventional semiconductors. However, how isotopic mass affects the optoelectronic properties in 2D semiconductors remains unclear because of measurement uncertainties resulting from sample heterogeneities. Here, we report an anomalous optical bandgap energy red shift of 13 (±7) milli–electron volts as mass of Mo isotopes is increased in laterally structured 100MoS2–92MoS2 monolayers grown by a two-step chemical vapor deposition that mitigates the effects of heterogeneities. This trend, which is opposite to that observed in conventional semiconductors, is explained by many-body perturbation and time-dependent density functional theories that reveal unusually large exciton binding energy renormalizations exceeding the ground-state renormalization energy due to strong coupling between confined excitons and phonons. The isotope effect on the optical bandgap reported here provides perspective on the important role of exciton-phonon coupling in the physical properties of two-dimensional materials.
This work was published in Science Advances.
[Y. Yu, V. Turkowski, J. A. Hachtel, A. A. Puretzky, A. V. Ievlev, N. U. Din, S. B. Harris, V. Iyer, C. M. Rouleau, T. S. Rahman, D. B. Geohegan, K. Xiao, “Anomalous isotope effect on the optical bandgap in a monolayer transition metal dichalcogenide semiconductor.” Sci. Adv. 10, eadj0758 (2024). https://doi.org/10.1126/sciadv.adj0758]
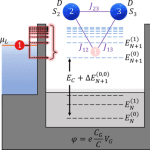
Mapping spin interactions from conductance peak splitting in Coulomb blockade
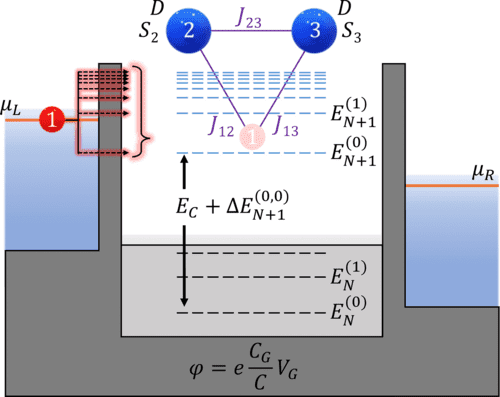
We investigate the transport properties of a quantum dot coupled to leads interacting with a multispin system using the generalized master equation within the Coulomb blockade regime. We find that if two states for each scattering region electron manifold are included, several signatures of the interacting spin system appear in steady-state transport properties. We provide a theoretical mapping of differential conductance peak signatures and all spin Hamiltonian parameters related to the inclusion of excited state transitions between uncharged and charged electron manifolds. Our predictions describe a scheme of only using a quantum dot and differential conductance to measure magnetic anisotropy, interspin exchange coupling, exchange coupling between the spin system and itinerant electron, and applied magnetic field response.
This work was published in Physical Review B.
[E. D. Switzer, X.-G. Zhang, V. Turkowski, T. S. Rahman, “Mapping spin interactions from conductance peak splitting in Coulomb blockade.” Phys. Rev. B. 108, 174438 (2023). https://doi.org/10.1103/PhysRevB.108.174438]
A closer look at how symmetry constraints and the spin–orbit coupling shape the electronic structure of Bi (111)
Fully relativistic density-functional-theory calculations of Bi(111) thin films are analyzed to revisit their two metallic surface-states branches. We first contrast these metallic branches with surface states arising at gaps in the valence band opened by the spin–orbit coupling (SOC). We find that the two metallic branches along Γ𝑀― do not overlap with the bulk band at the zone boundary, M. We show that the spin texture observed in such states cannot be traced to the lifting of Kramers’ degeneracy. Instead, we track them to the 𝑚𝑗=±1/2–𝑚𝑗=±3/2 SOC splitting, the potential anisotropy for in-plane and out-of-plane states, and the coupling between the opposite surfaces of a slab occurring near M, which is driven by a spatial redistribution of the four metallic states composing the two metallic branches. Each of these branches appears to be non-degenerate at the tested surface, yet each is degenerate with another state of opposite spin at the other surface. Nevertheless, the four metallic states bear some contribution on both surfaces of the film because of their spatial redistribution near M. The overlapping among these states near M, afforded by their spatial redistribution on both surfaces, causes a hybridization that perpetuates the splitting between the two branches, makes the film’s electronic structure thickness dependent near M, extinguishes the magnetic moment of the metallic states avoiding the magnetic-moment discontinuity at M, and denies the need or expectancy of the metallic branches becoming degenerate at M. We propose that the opposite spin polarization observed for the two metallic branches occurs because the surface atoms retain their covalent bonds and thus cannot afford magnetic polarization. We show that the Rashba-splitting of the metallic states for inversion-asymmetric films does not have a fixed magnitude but can be tuned by changing the perturbation breaking inversion symmetry.
This work was published in the Journal of Physics: Condensed Matter.
[M. Alcántara Ortigoza, T. S. Rahman, “A closer look at how symmetry constraints and the spin–orbit coupling shape the electronic structure of Bi (111).” J. Phys.: Condens. Matter 36, 015503 (2023). https://doi.org/10.1088/1361-648x/acfb67]
On stabilizing spin crossover molecule [Fe(tBu2qsal)2] on suitable supports: insights from ab initio studies
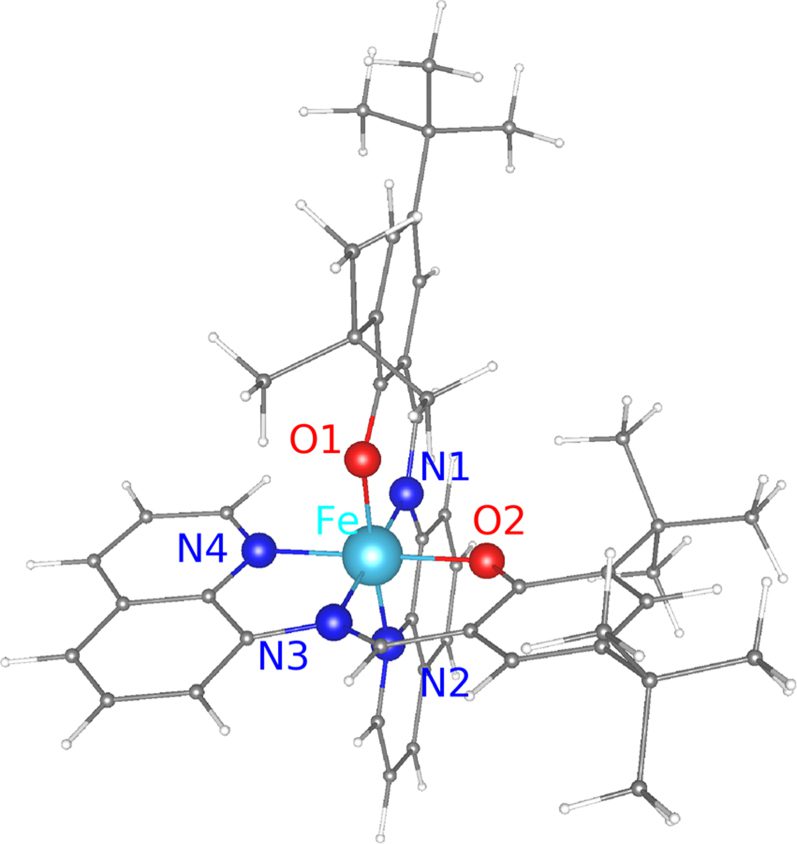
Au(111) is one of the substrates often used for supporting spin crossover (SCO) molecules, partly because of its inertness and partly because it is conducting. Using density functional theory based calculations of [Fe(tBu2qsal)2] SCO molecules adsorbed on the Au(111) surface, we show that while Au(111) may not be a suitable support for the molecule, it may be so for a monolayer (ML) of molecules. While, physisorption of [Fe(tBu2qsal)2] on Au(111) leads to electron transfer from the highest occupied molecular orbital to the substrate, electron transfer is minimal for a ML of [Fe(tBu2qsal)2] on Au(111), causing only negligible changes in the electronic structure and magnetic moment of the molecules. Furthermore, a small difference in energy between the ferromagnetic and antiferromagnetic configurations of the molecules in the ML indicates a weak magnetic coupling between the molecules. These results suggest Au(111) as a plausible support for a ML of [Fe(tBu2qsal)2], making such a molecular assembly suitable for electronic and spin transport applications. As for [Fe(tBu2qsal)2] SCO molecules themselves, we find hexagonal boron nitride (h-BN) to be a viable support for them, as there is hardly any charge transfer, while graphene displays stronger interaction with the molecule (than h-BN does) resulting in charge transfer from the molecule to graphene.
This work was published in Journal of Physics: Condensed Matter.
[D. Le, T. Jiang, M. Gakiya-Teruya, M. Shatruk, and T. S. Rahman, “On stabilizing spin crossover molecule [Fe(tBu2qsal)2] on suitable supports: insights from ab initio studies,” Journal of Physics: Condensed Matter 33, 385201 (2021). https://doi.org/10.1088/1361-648x/ac0beb]
Asymmetric Design of Spin-Crossover Complexes to Increase the Volatility for Surface Deposition
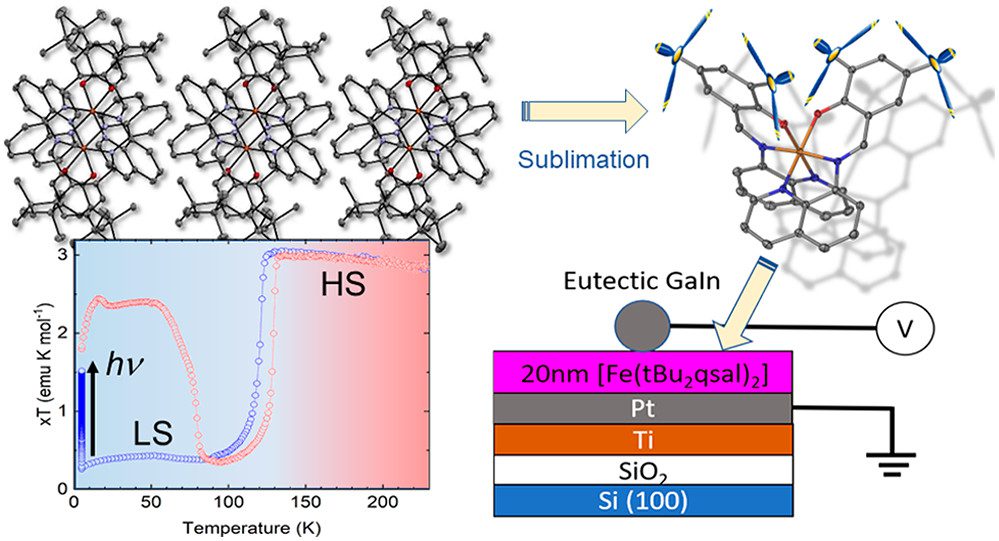
A mononuclear complex [Fe(tBu2qsal)2] has been obtained by a reaction between an Fe(II) precursor salt and a tridentate ligand 2,4-di(tert-butyl)-6-((quinoline-8-ylimino)methyl)phenol (tBu2qsalH) in the presence of triethylamine. The complex exhibits a hysteretic spin transition at 117 K upon cooling and 129 K upon warming, as well as light-induced excited spin-state trapping at lower temperatures. Although the strongly cooperative spin transition suggests substantial intermolecular interactions, the complex is readily sublimable, as evidenced by the growth of its single crystals by sublimation at 573 → 373 K and ∼10–3 mbar. This seemingly antagonistic behavior is explained by the asymmetric coordination environment, in which the tBu substituents and quinoline moieties appear on opposite sides of the complex. As a result, the structure is partitioned in well-defined layers separated by van der Waals interactions between the tBu groups, while the efficient cooperative interactions within the layer are provided by the quinoline-based moieties. The abrupt spin transition is preserved in a 20 nm thin film prepared by sublimation, as evidenced by abrupt and hysteretic changes in the dielectric properties in the temperature range comparable to the one around which the spin transition is observed for the bulk material. The changes in the dielectric response are in excellent agreement with differences in the dielectric tensor of the low-spin and high-spin crystal structures evaluated by density functional theory calculations. The substantially higher volatility of [Fe(tBu2qsal)2], as compared to a similar complex without tBu substituents, suggests that asymmetric molecular shapes offer an efficient design strategy to achieve sublimable complexes with strongly cooperative spin transitions.
This work was published in Journal of the American Chemical Society.
[M. Gakiya-Teruya, X. Jiang, D. Le, Ö. Üngör, A. J. Durrani, J. J. Koptur-Palenchar, J. Jiang, T. Jiang, M. W. Meisel, H.-P. Cheng, X.-G. Zhang, X.-X. Zhang, T. S. Rahman, A. F. Hebard, and M. Shatruk, “Asymmetric Design of Spin-Crossover Complexes to Increase the Volatility for Surface Deposition,” Journal of the American Chemical Society 143, 14563-14572 (2021). https://doi.org/10.1021/jacs.1c04598]
Group News
- New Publication: Increased Selectivity in Photolytic Activation of Nanoassemblies Compared to Thermal Activation in On-Surface Ullmann Coupling

- New Publication: Breaking Continuously Packed Bimetallic Sites to Singly Dispersed on Nonmetallic Support for Efficient Hydrogen Production
- Three Group Members Selected for Advanced School on Many-Body Physics
- New Publication: Anomalous isotope effect on the optical bandgap in a monolayer transition metal dichalcogenide semiconductor
- New Publication: Mapping spin interactions from conductance peak splitting in Coulomb blockade