
Computational physics (CP) is an approach to physics that uses computers to solve problems where a precise theory exists but the resulting equations are intractable to traditional analytical approaches. This area is relatively new in physics, but continues to grow in relevance especially as computational power and algorithms evolve. Computational approaches are applied to a wide range of areas, including condensed matter, surface physics, materials physics, soft-condensed matter, and biological physics.
Several groups in the UCF physics department make heavy use of computational approaches to address a wide array of physical problems:
Dr. Luca Argenti group:
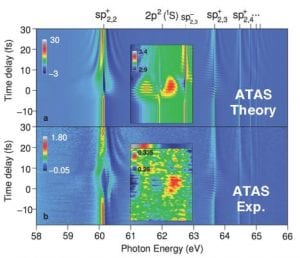
The group of Theoretical Attosecond Spectroscopies led by Dr. Luca Argenti develops new ab initio methods for the calculation of the electronic continuum of poly-electronic atoms and molecules to resolve photoemission in time, with the ultimate goal of controlling charge transfer processes in correlated electronic matter. To do so, it is necessary to solve the time-dependent Schrödinger equation and the Lippmann-Schwinger equation in a large configuration space to describe the evolution of excited electronic wave packets in time and extract from them the information they deliver asymptotically at the detectors. These calculations, which require the use of extensive numerical resources, allow one with interpret the rich observables made accessible by the new light sources, such as attosecond pump-probe laser setups and seeded x-ray Free-Electron Lasers.
Dr. Aniket Bhattachaya group:
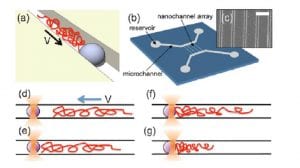
Dr. Bhattacharya uses scaling theory of polymer physics and a variety of numerical methods to study conformations and dynamics of DNA and other biopolymers under confinement. Most recent activities include investigations of DNA transportation through protein pores, confinement and non-equilibrium dynamics of DNA inside nano-channels.
Recent Publications:
- Kaifu Luo, Tapio Ala-Nissila, S-C Ying, and Aniket Bhattacharya, “Sequence dependence of DNA translocation thorough a nanopore”, Phys. Rev. Lett. 100 1058101 (2008).
- Aiqun Huang, H.-P Hsu, Aniket Bhattacharya, and Kurt Binder, “Semiflexible macromolecules in quasi-one-dimensional confinement: Discrete versus continuous bond angles”, J. Chem. Phys. 143, 243102 (2015). (Invited article)
- Aiqun Huan, Walter Reisner, and Aniket Bhattacharya, “Dynamics of DNA Squeezed inside a Nanochannel via a Sliding Gasket” (invited) Polymers 2016, 8(10), 352; doi:10.3390/polym8100352.
Dr. Abdelkader Kara group:
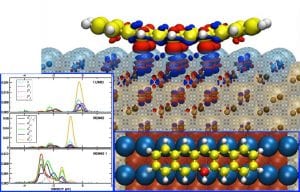
Fundamental understanding of the physical and chemical characteristics at surfaces and interfaces form the core of my computational research and development. Dr. Kara adopts a multi-scale/multi-tool approach to tackle seamlessly phenomena at different scales and environments. Of interest are the characteristics of the interface between organic materials and metal surfaces as they determine the performance of organic devices, which are becoming the devices of the future. Developing new computational tools incorporating artificial intelligence, machine learning and other state of the art methods is also an active line of research in the group.
Dr. Talat S. Rahman group:
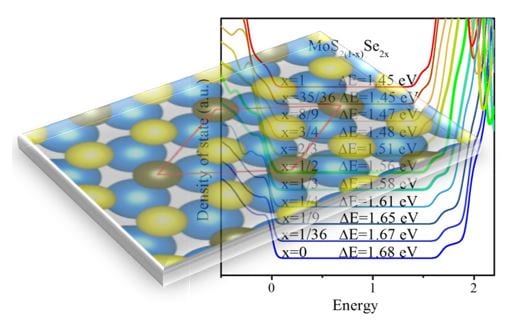
Our research is focused on theoretical and computational modeling of materials with particular interest in understanding mechanisms that control epitaxial growth and morphological evolution, chemical reactivity of nanostructured surfaces and nanoparticles, optical and electrical properties, with focusing on excited states, of materials. Very recently we have also ventured into developing the tools for understanding and modeling the properties of such biomaterials as peptides and proteins. The importance of this field is both technological (thin-film growth, nanotechnology for drug delivery, novel materials, catalysis, corrosion, lubrication, etc.) and fundamental. It raises questions about the nature of the bonding between atoms and molecules in regions of low symmetry and complex local environment and of how this bonding is affected by the electronic structure, microscopic geometry, atomic coordination and elemental characteristics of the atoms and molecules comprising various systems. In addressing these and related questions about the electronic structure, a distinct and important aspect of our work is also to probe the temperature dependencies of the properties, as accurately as feasible, so as to understand the behavior in laboratory environments. To achieve this our goal is to develop the framework for multi-scale modeling of materials in which comprehensive understanding developed at the atomic scale provides input parameters and physical insights for further examination of systems at larger length- and time-scale (in the mesoscopic range). Such studies have considerable predictive power and are expected provide the knowledge base necessary for tailoring functional materials by design.
Dr. Patrick Schelling group:
In this group, computational approaches including molecular-dynamics with empirical potentials and density-functional theory (DFT) are used to describe the coupled transport of heat and matter in solids and liquids. More recently Dr. Schelling has focused on the application of theoretical and experimental methods to understand the evolution of minerals relevant to planetary science. For example, the group is using empirical potentials based on the ReaxFF approach, developed and validated with DFT calculations, to describe how surface chemistry and defects relate to dissipation mechanisms in collisions between mineral grains, which is a key step in planet formation.
Dr. Sergey Stolbov group:
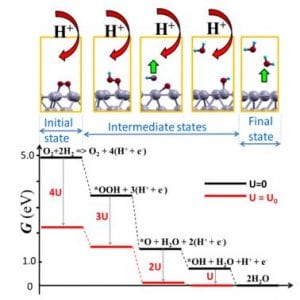
The main focus of the group is on rational computational design of the cost-effective stable and active electrocatalysts for the hydrogen fuel cell electrodes. The approach is as follows: a) preselection of eight to ten promising materials based on the existing knowledge of the relation between the catalyst surface composition, its electronic structure, stability and reactivity; b) first principle “testing” of stability of the materials and the thermodynamics of a reaction in question to narrow down the selection set to one or two most promising surfaces; c) experimental testing of the latter. We perform the search among atom monolayers or sandwiches of active elements on inexpensive substrates, as well as metal doped defected graphene.